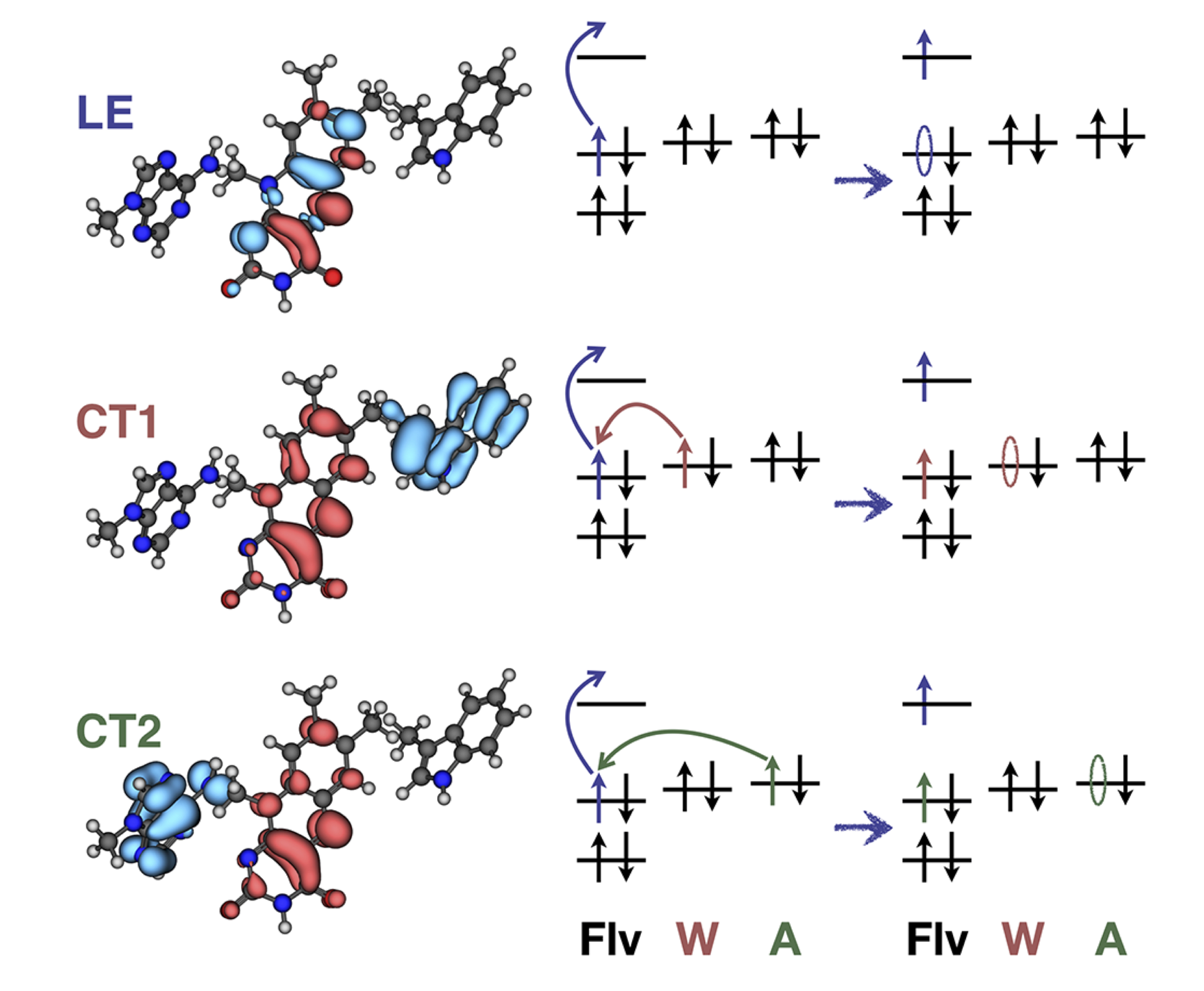
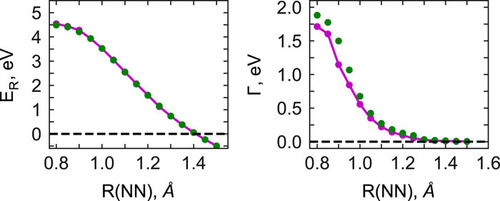
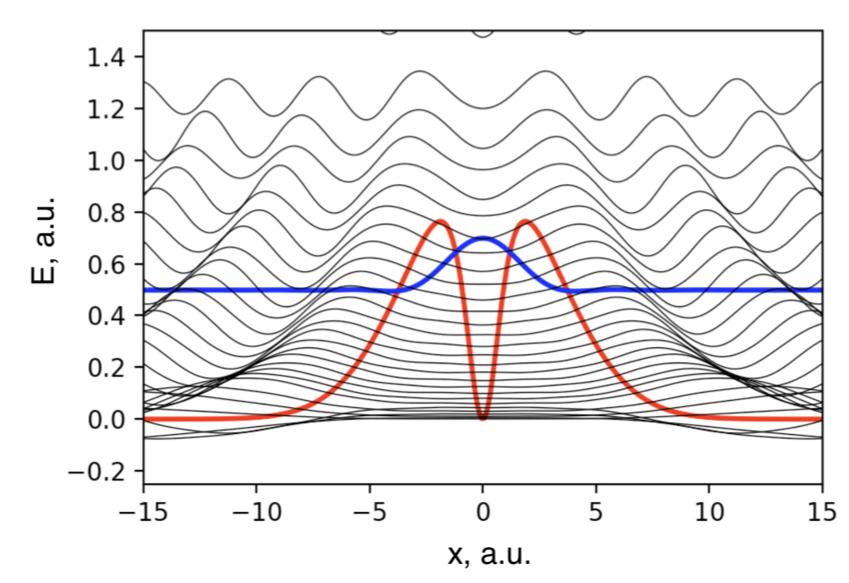
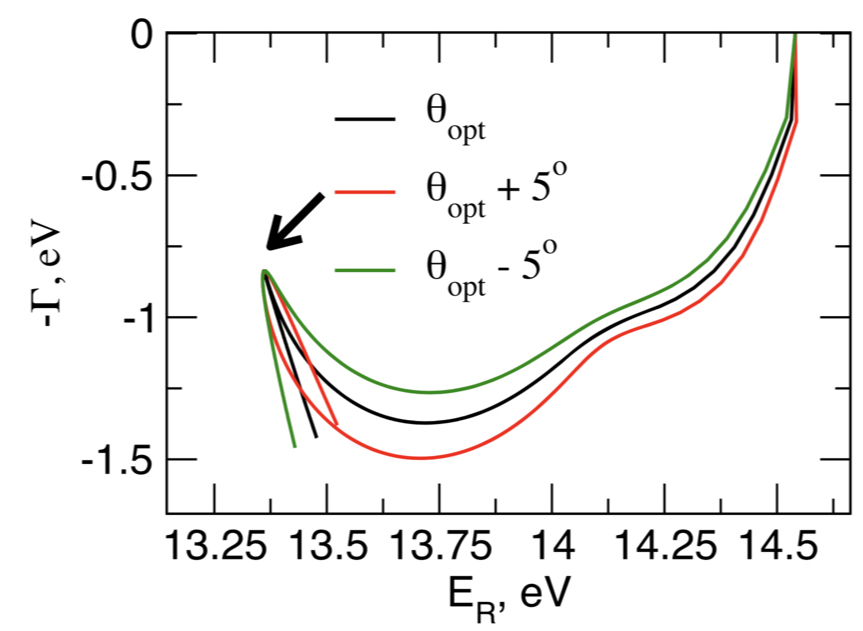
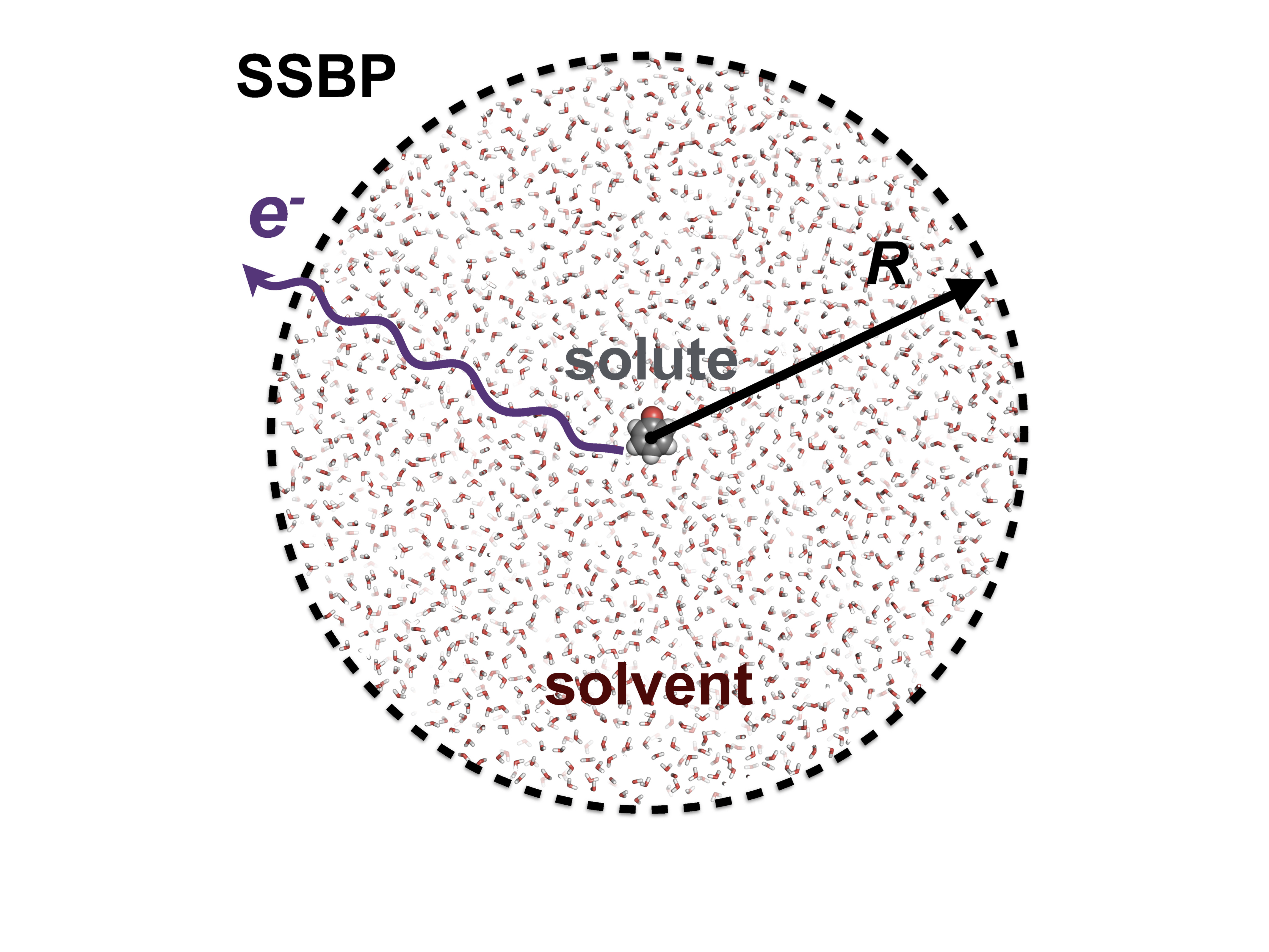
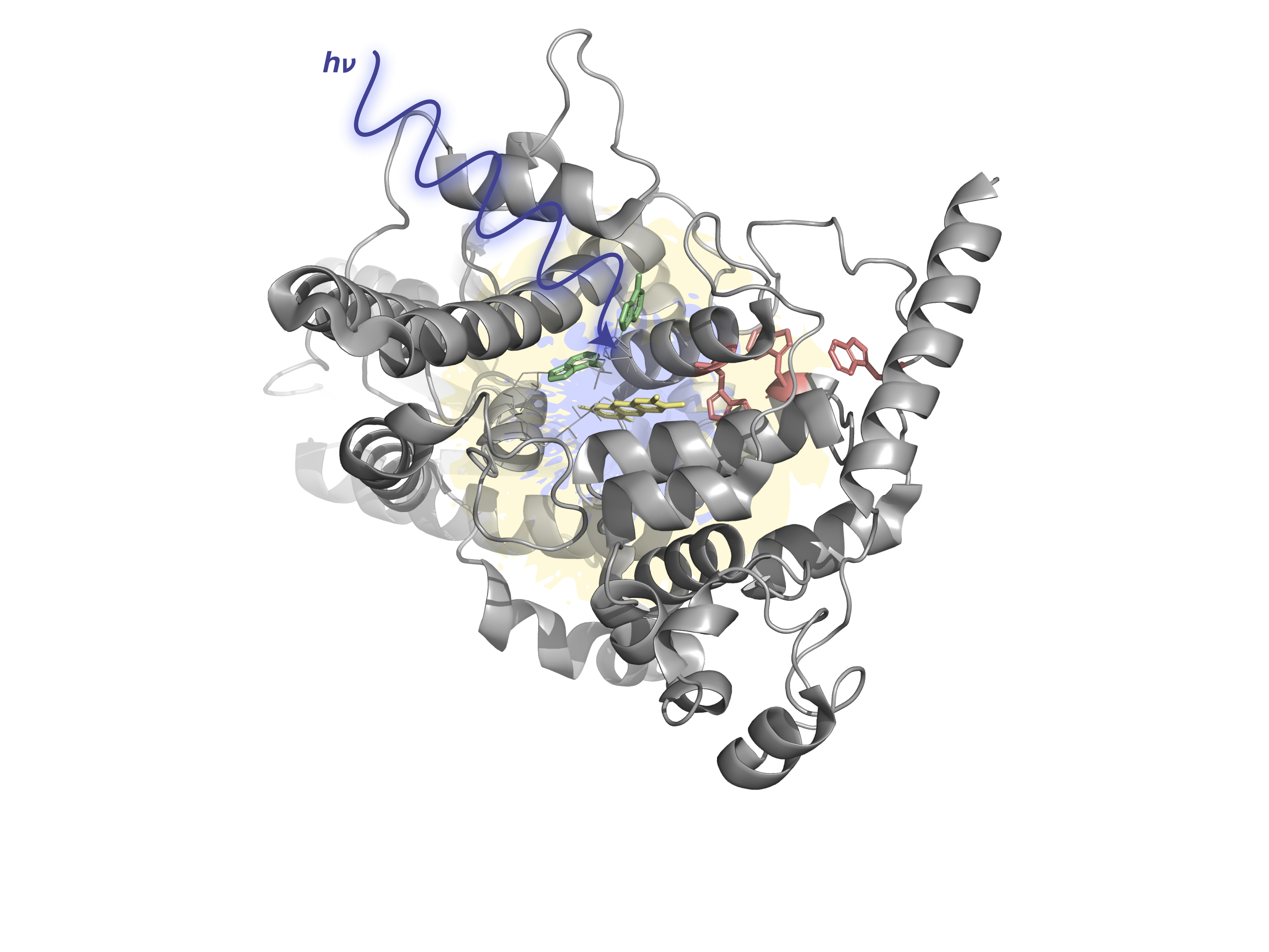
Metastable electronic states are states that lie in the autodetachment continuum, i.e. that have enough energy to eject an electron, and, yet, live long enough to be experimentally detected or even cause a chemical reaction. Metastable states, or electronic resonances, are commonly formed upon electron capture by molecular system or though excitation into highly-excited states. They serve as intermediates in such processes DNA building blocks radiation damage, are routinely formed in highly energetic environments, and possibly act as gateway states for formation of stable anions in the interstellar medium.
As resonances belong to the continuous spectrum of the Hamiltonian, they cannot be described using conventional quantum-chemistry methods. We develop new methods for describing energies and lifetimes of these states by combing Non-Hermitian quantum mechanics with advanced quantum chemistry electronic structure methods. We also exploit developed and existing techniques to study electronic structure of metastable electronic states and mechanism of electron capture by molecules. Several representative references on theory development and applied computational studies are listed below.